
Next-generation sequencing (NGS) has become a cornerstone of modern diagnostic workflows, including in hematological oncology, as it offers expansive information about samples’ genomic profiles. However, to harness its full potential, laboratories must balance two important factors: sequencing depth of coverage and sample batching.
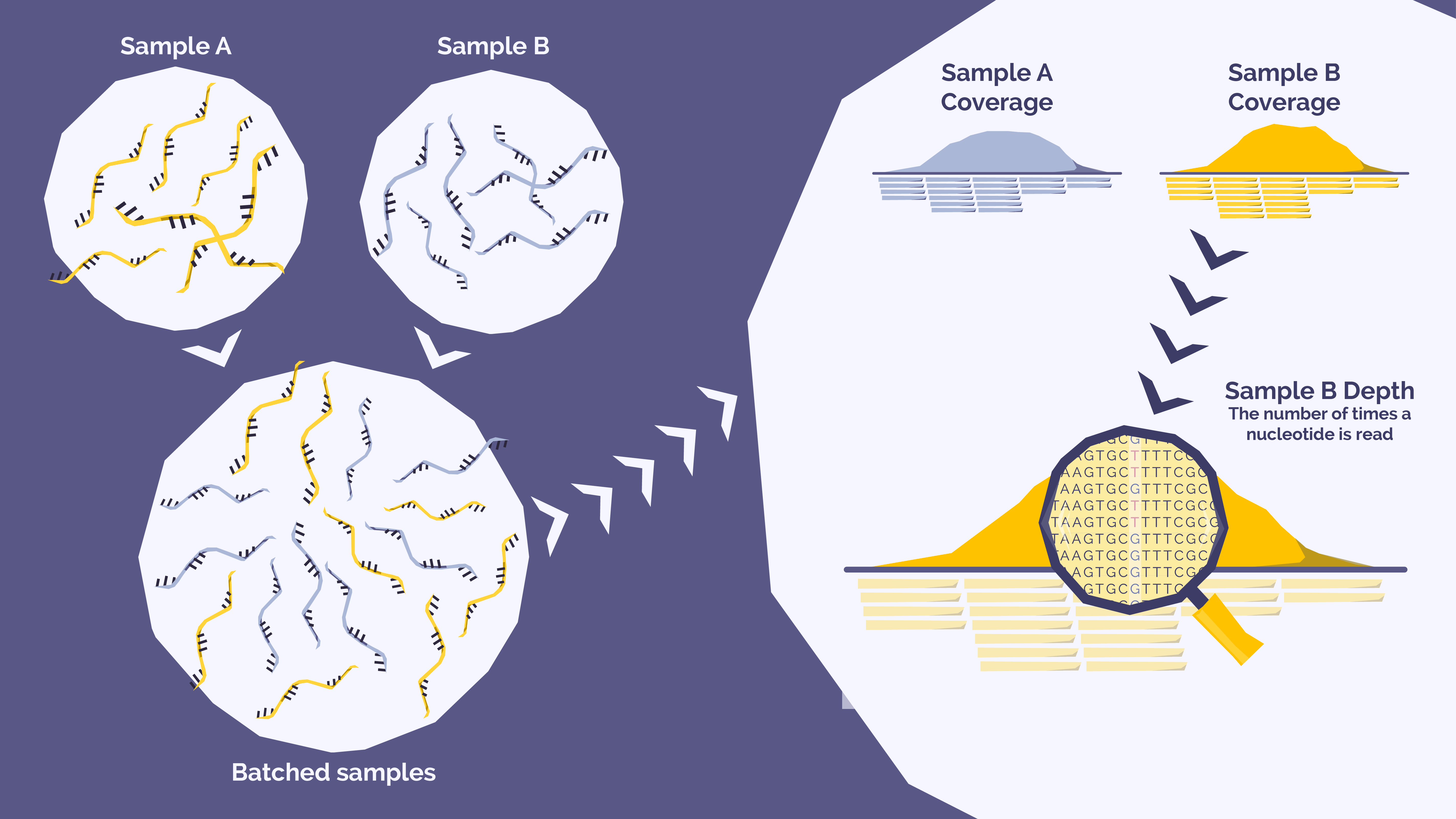
In hematological oncology, where the detection of rare variants (such as measurable residual disease or emerging clonal populations) which often have low variant allele frequencies (VAF, the proportion of sequence reads that contain a specific variant relative to the total number of reads at that position) is often critical, the balance between depth and batch size is a key consideration.
Sequencing depth is the number of times a nucleotide is read during a sequencing run and the mean target coverage depth (MTC) is the average number of reads for a specific targeted region; for example, an MTC of 1000x means that on average, that region in your sample has been sequenced 1000 times.
The limit of detection for NGS sequencing, and thus the capability to detect lower frequency variants, is directly related to the depth of sequencing performed. For example, if we consider a cancer sample where most of the cells are malignant (and thus carry the disease-associated variant at a high VAF), we can assume that a moderate sequencing depth would detect this variant. However, in cases such as measurable residual disease (MRD) testing, where variants may only be present in a very small fraction of cells (i.e. low VAF), a high sequencing depth is required to confidently detect these low frequency variants.
Several factors are relevant to depth requirements, including:
Sequencing batching is a strategy whereby multiple samples are pooled after preparation to be sequenced simultaneously. For example, multiple genetic samples may each be tagged with unique identifiers during the library preparation process. Samples can then be pooled together and sequenced on the same sequencing batch. The unique molecular identifiers are then used to differentiate samples after sequencing using a bioinformatic pipeline for analysis and differentiation of sample information.
This approach can potentially offer labs cost and time efficiencies by maximizing the use of a sequencing instrument’s capacity, reducing per-sample costs and user hands-on time as well as increasing sample throughput. But we must consider that a sequencer’s total capacity will be divided among those pooled samples. If too many samples are batched together, each sample may receive insufficient depth for reliable variant detection.
The impact of batching and depth is relatively straightforward:
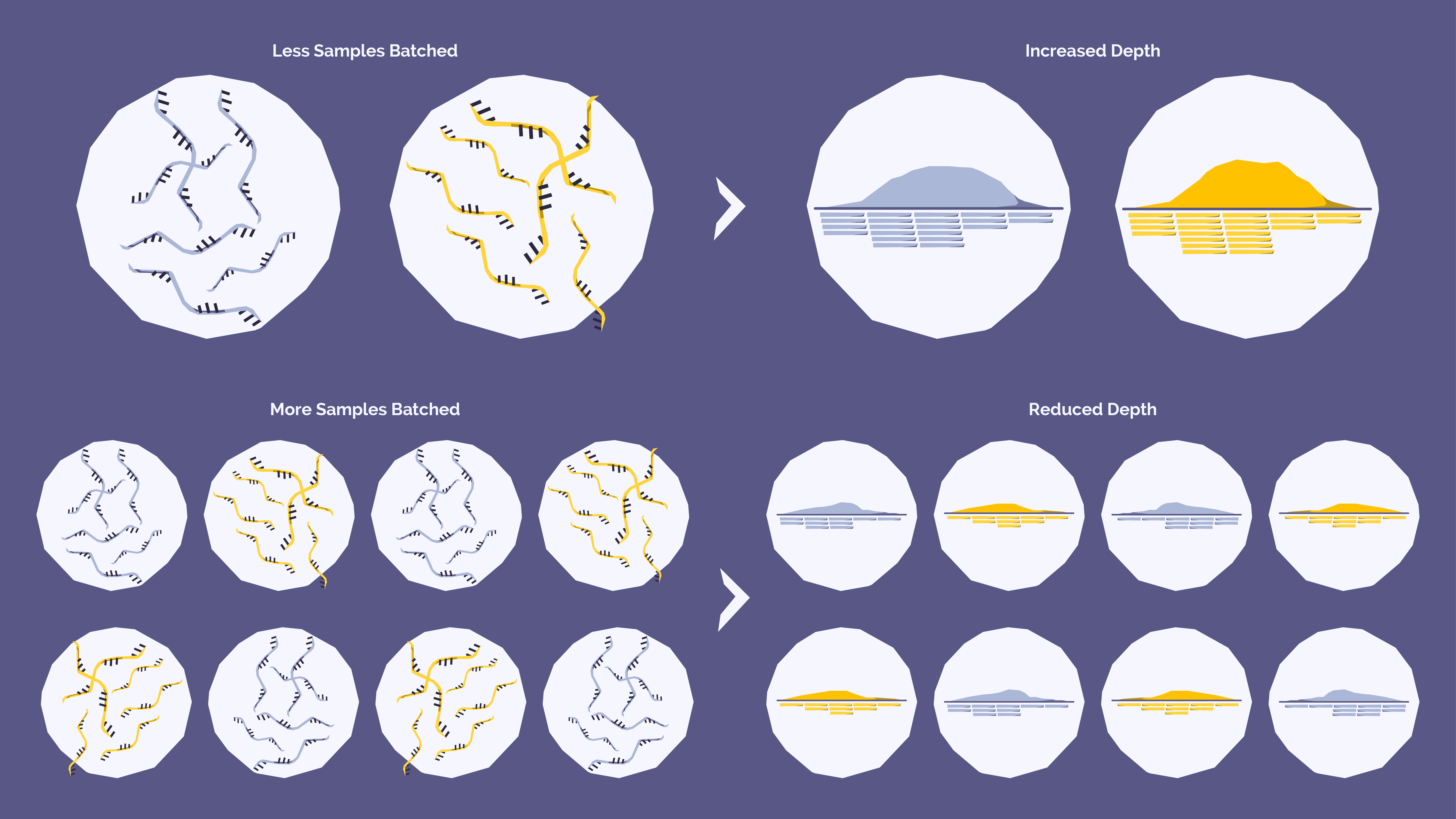
In hematological oncology, where the detection of rare variants can be clinically significant, balancing sequencing depth with batching strategies can be challenging. While the theory behind sequencing depth and batching is clear, practical implementation in a laboratory involves several additional considerations.
(*however this can be dependent upon flow cell capacity)
Through careful planning and ongoing optimization, laboratories can ensure that their NGS protocols are not only cost-effective but also sufficiently sensitive to detect subtle yet critical genomic changes. By understanding and applying these principles, you can design NGS workflows that are robust, sensitive, and tailored to the unique challenges of hematological oncology genomics.
View our range of SureSeq hematological malignancy NGS panels
Call +44 (0)1865 856800 Email contact@ogt.com
Send us a message and we will get back to you
 Visit International site
Visit International site Visit USA site
Visit USA site